| The first article is related to lipolysis and non-shivering thermogenisis. To not complicate things too much, I will stick to discussing only the local effects on fat cells and their adjacent nerves in this first instalment. |

Part 1: Lipolysis & Non-Shivering Thermogenesis
http://www.bodybuilding.com/fun/losefatnow.htm
By: Big Cat
This is the first article in a new series that will discuss, principally, the physiology of fat loss. The goal is to make you understand how fat loss occurs, and then evolve into a discussion on various supplements and drugs and how to best incorporate them (or not) into your supplementation scheme.
Although as always I will try my best to make things as understandable as possible, I can already tell you, if you are not familiar with physiology, you will have to be attentive in order to follow. The reward of reading this series can be enormous, as it will allow you to form a better understanding of why certain fat loss preparations work and others don't and allow you to make better choices with regards to diet and supplementation.
I suggest therefore that you grab a pen and paper as you read, and try to schematically represent what is outlined in this article. It involves a great deal of different enzymes and proteins and their abbreviations. They will not only be important in understanding this article, but also those that follow. So this is no excess luxury. When rereading the article, with your drawing, you will have much greater insight in how these processes work.
The first article is related to lipolysis and non-shivering thermogenisis. This is essential to make you understand the difference between white and brown adipose tissue, and still see the similarity in how they operate. To not complicate things too much, I will stick to discussing only the local effects on fat cells and their adjacent nerves in this first instalment.

Brown Adipose Tissue & White Adipose Tissue
There are two main types of fat deposits in the body. White adipose tissue is the unsightly fat we are all aiming to get rid off. All subcutaneous fat is usually white adipose tissue (WAT). The main function of white adipose tissue is the storage of energy, mainly fatty acids, in the form of tricglycerides. Triglycerides are three fatty acids, esterified to a glycerol backbone. In this form it is hard for them to escape the cell, and thus an ideal manner to be stocked.
In times of starvation, this white adipose tissue can then produce fatty acids for combustion in the liver and muscles (since fatty acids contain twice as much energy as glucose) which leaves the remaining glucose on your low calorie diet for the organs that cannot function without it, namely the brain and the sexual organs. Under stimulation of cathecholamines (adrenaline, noradrenaline) WAT will initiate a cascade that releases fatty acids from their glycerol backbone, allowing them to enter circulation when needed where they can be transported to the cells in need of energy.
Brown adipose tissue (BAT) on the other hand serves thermogenesis as its main purpose. Thermogenesis is the production of heat. BAT functions mainly in a similar manner as WAT, but it contains more stable Fatty acid Binding proteins (FABP) that keep the released fatty acids inside the cell. Unlike WAT, BAT is also metabolically active, meaning it can use energy itself.
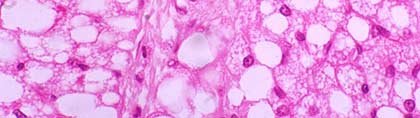
(Click To Enlarge) Brown fat magnified. In this case it will use its fatty acids and burn them, but at the same time 'uncouple' the production of ATP (the main form of energy in the body) from the combustion of its fatty acids. This makes the production of energy less efficient and leads to the production of heat instead. Normally heat is a by-product of energy production, in this case the cell deliberately makes its production less efficient to produce more heat, creating thermogenesis. It is therefore theorized that BAT played an important role in the survival of several mammal species by being able to produce extra heat in times of cold.
Making the distinction between WAT and BAT is extremely important to follow later follow-up articles on fat loss supplementation. Mainly because of the differences involved in their ways of contribution to fat loss. For instance understanding that when BAT takes up fat, it can be a good thing, since it can do so under stimulation of cathecholamines and thus absorb fat released from WAT and burn it, while uptake of fat by WAT generally leads to fat gain.
The regulation of the two also occurs in a slightly different manner. As we will see BAT is deficient in the beta2 adrenergic receptor and rich in the beta3 adrenergic receptor, while WAT is rich in beta2 adrenergic receptors, but has relatively few beta3 receptors. We will cover the significance of this in following paragraphs.
The Adrenoreceptors
The functioning of adipose tissue is regulated by adrenoreceptors and also mediated by other receptors, such as the adenosine receptors. The main effector of the adrenoreceptors is norepinephrine or noradrenaline (NE). In this article we will discuss the effect on fat cells, so I will limit myself to the actions on the fat cell and the adjacent nerve. The nerve release NE which then binds to the adrenoreceptors. NE is a ligand for all adrenoreceptors.
We will distinguish three main categories:
1. The beta adrenoreceptors (BAR): There are three BAR's, numbered 1 to 3. The B2AR is most commonly expressed in most cell types. With regards to adipose tissue we will see that it is however absent in BAT (8). BAT is mainly regulated by the B3AR. The B3AR is unique to adipose tissue and is highly expressed in BAT, and to a much lesser extent in WAT.
So we can already distinguish that the B3AR will predominantly relate to BAT, while the B2AR will predominantly relate to WAT. There is also a B1AR, which is of lesser importance. It most likely mediated the adrenergic response under normal conditions, because a lot of the processes mediated by the other two will continue in animals that do not express those receptors. BAR's increase lipolysis and thermogenesis and are the main players in these processes.
The way the BAR's work is as follows: The receptors are located on the outside of a cell. When a suited ligand (NE for example) binds to it, it activates a G-protein located on the inside of the cell membrane. A G-protein has three subunits, one of them, the alpha subunit, is attached to Guanosine Di-Phosphate (GDP). When the receptor activates the G-protein, the alpha subunit releases its GDP and binds GTP (guanosine Tri-phosphate). This causes the subunit to break away and activate an enzyme called adenylate Cyclase (AC, also called Adenylyl cyclase).
The function of this enzyme is to turn the cell's ATP into cyclic AMP (cAMP). cAMP is called the second messenger, it carries out the function of the first messenger (in this case NE) inside the cell. cAMP does a number of things, including the activation of exchange proteins and cation channels, but most important to us in this discussion is its phosphorylation of Protein Kinase A (PKA). Phosphorylation is the process of adding a phosphate ion to the protein, thereby either activating or inactivating it. In this it activates PKA. The further functions and relevance of PKA will be discussed at a later point.
2. The alpha1 adrenoreceptor (A1AR):
The alpha1 receptor is also a lipolytic receptor but works in a different manner. It uses Calcium (Ca2+) as a second messenger instead of cAMP. It's also considerably less lipolytic than the BAR's, at most 1/10th of the activity. Most likely to maintain some basal functions of the fat cell under less lipolytic conditions.
Like BAR's the A1AR activates a G-protein, this G-protein then breaks down Phosphatodylinositol-4, 5-biphosphate (PIP2) into 1,4,5-inositol Triphosphate (IP3) and DiacylGlycerides (DG's). IP3 then mediates the release of Ca2+ from intracellular stores allowing it to act as a second messenger. The DG's lead to phosphorylation and activation of Protein Kinase C (PKC). Ca2+ and PKC are the main effectors of A1AR activity in adipocytes.
Sometimes BAR's and A1AR's fulfil similar functions. When this occurs, the effector is most likely Src. This protein is activated through PKC (4) and Ca2+, but also via PKA (5). Although the mediation via PKA should theoretically (not verified) involve some form of tyrosine kinase.
3. The alpha2 adrenoreceptor (A2AR):
 The alpha2 receptor is, in contrast to the other two types, an anti-lipolytic receptor. It inhibits fat loss, at least locally. It can contribute to fat loss centrally, via actions on the brain, but since we are only discussing the fat cell and its adjacent nerves, we will consider the A2AR an anti-lipolytic receptor.
The alpha2 receptor is, in contrast to the other two types, an anti-lipolytic receptor. It inhibits fat loss, at least locally. It can contribute to fat loss centrally, via actions on the brain, but since we are only discussing the fat cell and its adjacent nerves, we will consider the A2AR an anti-lipolytic receptor.
It's also anti-thermogenic, but I will not keep repeating that, since lipolysis is essential for BAT thermogenesis, one automatically implies the other. Here too the mechanism is related to the activation of a G-protein, like with the BAR's. It will also act on adenylate cyclase, but instead of activating it, it will shut down adenylate cyclase activity, thus reducing the activity of the BAR's.
The Adenosine Receptors
The adenosine receptors are also anti-lipolytic receptors, that reduce adenylate cyclase activity and inhibit cAMP accumulation. Adenosine is sort of a feedback mechanism. Its released from the fat cell itself when energy is low. When a high amount of ATP is used to form cAMP, then the ATP:AMP ratio will be low, signifying low energy.
A second factor in the release of adenosine may be the A1AR. Using Ca2+ as a second messenger it can increases levels and activity of phosphodiesterases (PDE). PDE cause the breakdown of cAMP to AMP further reducing the energy ratio and causing more release of adenosine from the fat cell.
Adenosine, once released attaches to its receptor and further inhibits cAMP accumulation. This is further validated by evidence that A1AR activity increases blood flow to the cell, but that this blood flow is not mediated by glycerol release. The only other factor I can think of that would increase blood flow in such a manner would be adenosine. This could explain why this receptor is only slightly lipolytic, in contrats to the BAR's.
Location Of Adrenoreceptors & Adenosine Receptors
All these receptors are expressed on the cell surface, obviously, of the fat cell and mediate their activity, lipolytic or anti-lipolytic, inside the cell via the use of second messengers. But these receptors are also located elsewhere in the fat tissue. For starters, both the A2AR and the adenosine receptor are present on the end-terminal of adjacent nerves.
This serves as a negative feedback signal. When NE is released from the nerve, it will bind to these inhibitory receptors on the nerve itself and stimulate the reuptake of NE. This causes less available NE and thus lowered lipolysis. These findings demonstrate that at least at a local level, the blockade of A2AR and adenosine receptors is a valid and versatile means of increasing lipolysis.
BAR's, and specifically the B2AR is also highly expressed in the vascular beds of the fat tissue, where it regulates blood flow. NE can both cause vasoconstriction and vasodilation in cardiovascular tissue, making it the prime regulator of blood flow to several organs and the distribution of fatty acids throughout the body. This further emphasizes the importance of stimulating BAR's to enhance lipolysis.
Regulation Of Lipolysis
When we say the A1AR is lipolytic, this deserves some nuance. Lipolysis is the process of releasing fatty acids from their glycerol backbone. While the A1AR supports fat loss in several fashions, it is not lipolytic per se. Lipolysis is namely regulated by Protein Kinase A, which is only activated via cAMP, and thus BAR's.
Protein Kinase A regulates lipolysis in a dual fashion. First it phosphorylates and activates Hormone sensitive Lipase (HSL). HSL initiates a three step catalytic process that releases a fatty acid from the triglyceride molecule in each step, yielding three Free Fatty acids (FFA) and glycerol.
Glycerol is free to flow out of the cell and since it is highly hygroscopic (attracts water) it will improve blood flow to the cell. If the FFA's can be transported out of the cell, as is often the case in WAT, it will therefore facilitate their systemic uptake.
However it seems that triacylglycerol (TAG) is quite resistant to HSL, because it is surrounded by perilipin (1). That is the second fashion in which PKA will improve lipolysis, namely by phosphorylating and deactivating perilipin, freeing up the TAG that is now more susceptible to breakdown by HSL.
Lipolysis in WAT is the primary goal, since now we have FFA's that can be transported out of the cell and used systemically to be combusted for energy. This causes a reduction in WAT size and this is what we are aiming for, to lose that ugly fat.
Understandably these processes do not occur when you are taking in a lot of food since then there will never be a call for fat tissue to release FFA's. And likewise, if you manage to stimulate the release of FFA's but you are eating too much, they simply will not be burned and be re-esterified.
Lipolysis Leads To Thermogenisis In BAT
 As we discussed earlier BAT is metabolically active and induces mitochondrial uncoupling to produce heat. Since the body still needs the same amount of energy, a reduction of metabolic efficiency leads to a greater need in calories.
As we discussed earlier BAT is metabolically active and induces mitochondrial uncoupling to produce heat. Since the body still needs the same amount of energy, a reduction of metabolic efficiency leads to a greater need in calories.
When thermogenesis occurs, it is therefore beneficial to fat loss since you burn more fat for a given amount of food taken in. Mitochondrial uncoupling is induced by an uncoupling protein, UCP1 (also called thermogenin).
So in BAT, lipolysis is not the final step, but increased expression and activation of UCP1 is. Expression of UCP1 is regulated by both BAR's and A1AR.
This suggests that this occurs through Src. Src is capable of activating two (5,6) of the three mitogen activated protein kinasas (MAPK) as well, and since one of them (p38 MAPK) has been named in expression of UCP1 (6) it is likely that it occurs through this Kinase.
However A1AR mediated increase in UCP1 expression is not completely blunted by inhibition of Src, suggesting a direct mediation by PKC as well. However to what extent is not known. Since both A1AR and BAR's were found to be equipotent in their induction of UCP1 expression its likely that Src is the main regulator.
In any case, the final step is the phosphorylation and activation of CREB (7), which directly increases UCP1 mRNA in the cell. It also produces ICER, a negative regulator of CREB, as a negative feedback signal. With this increased expression of UCP1, possibility of activation is insured. The actual activation of UCP1 however occurs under the influence of lipolysis, or rather the presence of Fatty acids.
Some FFA's are taken up by Fatty acid binding proteins (FABP). BAT expresses more stable FABP than WAT does, so most of the FFA's end up bound and remain in the cell. Even though BAT can produce FFA's, it does not do so to a great extent. Instead the FA content in the cell leads to activation of UCP1 and results in the burning of the fat and the production of heat.
Why Is This Important?
Many may reason that its only using its own FA's and thus does not contribute to fat loss. But that is not the case. In BAT, NE stimulation via a cAMP dependent manner, increases Lipoprotein Lipase (LPL) expression, which leads to the uptake of FA's and triglycerides from the blood.
In contrast, NE will lead to downregulation of LPL in WAT. So the same stimulus that causes the release of FFA's from WAT, causes the uptake of fats into BAT for burning. This makes BAT a useful partner is the fat burning process, but in a totally different manner than WAT. We don't really want BAT to atrophy, on the contrary, we want it to take up as much fat as it can during the diet.
Thermogenesis can also occur in other tissues, to a much lesser extent. Including in WAT. However this thermogenesis is not dependent on UCP1 (3), since UCP1 is only expressed in BAT (2).
Conclusions
This is a lot of information to process all at once. No doubt the different cascades and the involvement of all these enzymes and proteins have confused you somewhat. I hope you took my advice and drew it out schematically. When you reread the article with the drawing next to you, you will be able to grasp all the concepts discussed here more clearly.
All of this will be of extreme importance in understanding the following articles in these series, so I do hope you take the time to let these things sink in. It will be worth your while in understanding fat loss, and the best mechanisms to manipulate it in the end so you can achieve maximum fat loss in a minimum amount of time, with little or no loss of muscle.
In any case we have already learned various ways of manipulation that will lead to increased fat loss. The stimulation of the beta adrenoreceptors and of the alpha1 adrenoreceptor, and all their downstream targets. The blockade (at least locally) of the alpha2 adrenoreceptor and the adenosine receptors and of course the reduction of PDE produced by the A1AR.
Part 2: Insulin Resistance & The Modulation Of PPARgamma!
I hope you have had the time to reread the first article and comprehend it fully before starting here. This article will be considerably simpler to understand than the previous one, but only truly so if you fully comprehend the first article. This time around we will discuss insulin resistance and how it works to our advantage in various ways, as well as the manipulation of the first of the PPAR's, PPARgamma, which I consider to be a vital part in proper fat loss.
Understanding The Relevance Of Insulin Resistance In Fat Cells
There are a lot of misconceptions about the relevance of insulin resistance. Some people believe that increased insulin sensitivity is a valid approach to fat loss. This originates largely from the idea that obesity is often the cause of extreme insulin insensitivity. Therefore in these people, and increase in insulin sensitivity will lead to repartitioning of nutrients and may actually cause a reduction in body-fat.
 Most who read this however, I take it, are not exactly obese. In normal people insulin is the primary adipogenic (fat building) hormone. Therefor a reduction in insulin, and sensitivity of cells to insulin, is a positive thing for fat loss. Any fat loss preparation that contains insulin sensitizing agents is therefore not suited if your goal is to get really ripped. Products like ALA and r-ALA, free form L-taurine, D-Pinitol and the like are therefore absolute scams if they are being sold for fat loss purposes.
Most who read this however, I take it, are not exactly obese. In normal people insulin is the primary adipogenic (fat building) hormone. Therefor a reduction in insulin, and sensitivity of cells to insulin, is a positive thing for fat loss. Any fat loss preparation that contains insulin sensitizing agents is therefore not suited if your goal is to get really ripped. Products like ALA and r-ALA, free form L-taurine, D-Pinitol and the like are therefore absolute scams if they are being sold for fat loss purposes.
Insulin activates several enzymes needed for fat gain, such as Fatty acid synthase (FAS) etc. It also increases glycogen stores. While this is a good thing for performance, it is a negative aspect for fat loss. When norepinephrine (NE, cfr part1) is released it will begin by acting as a glycolytic agent, that means it turns glycogen back to glucose. At this point that means the metabolically active cells are still using glucose since it is readily available, instead of burning fat.
I won't discuss this in detail, as we will revisit this aspect in the third instalment, but when glucose becomes low, ATP:AMP ratio (energy balance) drops and that activates AMPK, a Kinase that tells the mitochondria to burn fat for energy instead of glucose. At this point the FFA's produced by WAT (cfr part 1) can be burned).
Most bodybuilders are already aware of this, since they will not only lower calories during a diet, but lower mainly carbohydrates from the diet. This is because carbohydrates increase blood glucose and blood glucose elevates insulin. So in essence you already know that less insulin equals more fat loss.
What is however important to understand is that a reduction in insulin signalling means a reduction in protein synthesis, and thus, later in the diet, a greater chance of muscle loss as body-fat gets lower. We must therefore distinguish between insulin resistance in the fat cell and insulin resistance in the muscle cell. Both will promote fat loss.
But if we can promote insulin resistance in the fat cell without promoting it in the muscle cell, we may take a little longer to lose body-fat, but we might spare more muscle. The reason insulin insensitivity in muscle helps fat loss is the storage of glycogen. This would however not be a major factor when dieting with low calories and low carbs. First of all there wouldn't be much glucose to store as glycogen, and secondly the low glucose level would keep insulin levels down as well.
Norepinephrine Inhibits Insulin Release
From part 1 of this article series, it should already become clear that increasing the release of NE will be crucial in stimulating maximal fat loss. And we haven't even discussed its effects on muscle retention yet.
Well, NE may play multiple roles in this process via its inhibition of insulin. When NE release is increased, NE is also released near the beta cells in the pancreas, responsible for secretion of insulin, and inhibits the release of insulin. So again, insulin becomes less of a factor in fat loss.
Interleukin-6 Is Perhaps The Best Target
Another way in which NE aids in this aspect is by stimulating the release of Interleukin-6 (IL6). The release occurs through both PKA and Ca2+ (2) and is thus likely mediated by Src, possibly through its activation of one or more Mitogen activated protein Kinases (MAPK). IL6 is a particularly interesting target as well, because it seems to increase insulin sensitivity in muscle tissue, while it inhibits insulin sensitivity in adipose tissue (1).
In the fat cell an increase in IL6 will primarily result in a reduction of the cytokine adiponectin(4). Adiponectin is known to improve insulin sensitivity. So by inhibiting its release, IL6 makes the fat cell less sensitive to insulin.
At the same time it seems to activate the Supressor of Cytokine Signalling (SOCS3), also activated by insulin itself, which would reduce insulin sensitivity. This is one of the negative feedback signals for insulin signalling as well. Combined a fat cell would experience an immense reduction in sensitivity to insulin.
This becomes evident in the reduced expression of mediators of insulin-related fat gain, such as Fatty Acid Synthase (FAS) and acetyl-CoA carboxylase, and of the transcription factors C/EPBalpha and PPARgamma (which will be discussed at length later on). IL6 is therefore one of the most promising targets in both fat loss and muscle gain. This is also supported by the fact that IL6 is upregulated by physical activity, improving muscle gain and glucose disposal in muscle, while reducing fat gain in fat cells by opposite mechanisms.
Norepinephrine May Reduce Insulin Sensitivity Via AMPK
As we saw in part one, and earlier in part 2, a negative energy balance leads to activation of AMPK. Increase NE will definitely lead to increase in AMPK activation. First of all the activation of Adenylate Cyclase (AC) via the BAR's will lead to ATP being broken down to make cAMP.
Secondly, via the A1AR and its second messenger, Ca2+, we experience an increase in PDE's that breakdown cAMP to AMP, further lowering the ATP:AMP balance. Whether or not activation of AMPK leads to insulin resistance is not entirely sure, but a number of insulin's adipogenic markers are definitely reduced during AMPK activation (5).
There is no established link between AMPK and IL6 (6), so they regulate their effects independently of each other and may be additive in their effects.
What Is PPARgamma?
PPARgamma stands for peroxisome proliferator activated receptor gamma. There are three PPAR's, nuclear receptors, that all modulate fat homeostasis. However the other two, the alpha and beta receptor, have a positive effect on fat loss, predominantly via effects on liver and muscle respectively, improving the oxidation of fatty acids. PPARgamma on the other hand is a negative regulator of fat loss (7).
It is a key factor in adipocyte (fat cell) differentiation, or if you will, the recruitment of more adipocytes. It also increases the synthesis of fatty acids and the uptake and storage of glucose (9) and downregulates the B3AR (8). And if you remember the inhibitory role of perilipin on lipolysis from the previous article, well PPARgamma increases perilipin (3). Inhibiting PPARgamma is therefore a valid, and not often enough explored, pathway for increasing fat loss.
In fat cells, a reduction in PPARgamma would reduce adipogenesis drastically, thus allowing for more fat loss and less fat gain. The role of PPARgamma in fat gain is easily demonstrated, as bodybuilders have in the past (and still do) used PPARgamma agonists like metformin, with as a result an enormous amount of fat gain over a short period of time.
Because PPARgamma plays a role in increasing insulin sensitivity, they assumed it would mimic the effects of insulin. As we saw at the beginning of part 2 however, for a non-obese, non-diabetic person, such methods are not well suited for fat loss and may have, obviously, the opposite effects.
In fat cells there are multiple mechanisms to reduce PPARgamma. Most of them likely related to a reduction in insulin sensitivity. IL6 for instance, lowers PPARgamma expression, and it has even been postulated that this is the manner in which it lowers adiponectin. Likewise, AMPK will reduce PPARgamma as well, independent of IL6. Manipulation of IL6 and AMPK is however not the end all of PPARgamma downregulation. We can also supplement with specific antagonists of the receptor. Which is something we will definitely discuss in later instalments of this series.
Part 3: Fatty Acid Oxidation, Whole Body Effects And Thyroid Hormone!
The bad new is, if you don't understand or haven't read the first two parts (part one and part two), you may be a little lost reading this article, since I make a few references. The good news is, however complicated this stuff may be at first glance, if you have read and comprehend the first two articles, then this one should be an absolute breeze. In this one I will try to tackle fatty acid oxidation, the whole body beneficial effects of alpha2 adrenoreceptor stimulation and the effects of thyroid hormone.
Fatty Acid Oxidation Is The Final Step In Optimizing Fat Burning
Fat loss has forever centered around lipolysis and reduction in appetite. However, reducing food intake and increasing the release of fatty acids is just one step, now you have to also make sure the released fatty acids actually get burned. In caloric deficit, on a low carb diet this will occur regardless. But that doesn't mean we can't help the process along a bit. In order to do this, we are going to discuss the mechanisms by which fatty acid oxidation increases.
Food stuffs get burned in metabolically active cells. These cells possess mitochondria, which you could portray as little factories that take your macronutrients and turn them into ATP for energy. Under normal circumstances, these little factories work on glucose. However there are organs in the body that rely on glucose more, such as for instance the brain.
ATP Molecule When a glucose shortage occurs, the glucose will be spared for the brain, and the other metabolic active tissues will have to function on something else. This is why we actually have fat. Fat is easily stored in the body and contains twice as many calories as glucose.
When we require energy but don't get enough from our food we initiate lipolysis to acquire fat as a substrate for energy production. This means the mitochondria must adapt to burning fat instead.
The main users of energy are the liver and the muscles. This also clarifies why bodybuilders have considerably less trouble losing fat than your average person, because we simply have a higher amount of metabolically active cells. The change in these cells occurs largely through processes we previously discussed in fat cells (cfr part2) regarding insulin resistance, namely through AMPK activation (as a result of low energy balance) or stimulation of Interleukin-6 (IL6).
Both these occurances have been shown to reduce the adipogenic (fat gaining) market acetyl-CoA carboxylase (ACC). To oxidize a fatty acid, you need to get it into the mitochondria, this occurs via carrier molecules, in this case Carnitine Palmitoyl tranferase (CPT) . What ACC does is create a product called malonyl CoA that basically inhibits CPT from actively carrying out its duty.
So when AMPK reduces ACC we are in fact freeing up more CPT that can transport fatty acids to the mitochondria. Again we are staying in the same line of manipulation.
PPARalpha And Beta
In the last article we also discussed a nuclear receptor called PPARgamma, and said it had two other brothers, namely the alpha and the beta receptor. These two exert positive effects by increasing fatty acid oxidation. PPARalpha agonists are widely used in the reduction of cholesterol. This is because they induce fatty acid oxidation in the mitochondria of the liver.
In the liver the beta receptor, however, is nothing more than a failsafe. In muscle tissue, the situation is the opposite. This tissue is quite poor in the alpha receptor and quite rich in the beta receptor. Here the beta receptor will play the crucial role (1).
The use of common PPARalpha agonists, such as fibrates, is sometimes recommended to increase fatty acid oxidation. But in the real world such a thing has failed miserably. A true explanation I couldn't give you, maybe because the induction of the alpha receptor already occurs readily in a starvation state. There is however one fibrate that seems to elicit minor successes, namely bezafibrate.
This is possibly because bezafibrate is also a PPARbeta stimulator (2). In bodybuilders it makes more sense to target the PPARbeta receptor anyway, since our bodies comprise of over (sometimes well over) 50% muscle mass. With that much metabolically active tissue, tuned to oxidize fatty acids, must evoke some type of increased reaction in fat loss.
When we get down to discussing various fat loss products, we will encounter more ligands for these receptors. But it is important to know that the body too makes an endogenous ligand for the PPARbeta receptor, namely prostacyclin (3,5) (Prostaglandin I2 or PGI2). Stimulation of BAR's by, for example, NE to stay in the same line, does lead to a modest increase in PGI2 production (4).
Whole Body Effect Of A2AR Stimulation
We first discussed the alpha2 adrenoreceptor (A2AR) in part one of this article series. We said it was an anti-lipolytic receptor, at least locally in the fat cells. Which implied that systemically it may produce some form of positive effect. This is most certainly the case. When stimulating the alpha2 receptor in the brain, part of its effects are a reduction in the orexigenic peptide Neuropeptide Y (NPY) (6).
Orixigenic implies that it stimulates appetite. Which means a reduction in NPY will reduce appetite. This will be important to remember, as in the next article we will discuss appetite suppression, and in the article after that why systemic blockade of the A2AR has proven a poor choice in fat loss.
But NPY apparently does more than just regulate appetite, it exerts a profoundly negative effect on thyrotropin Releasing hormone(7) (TRH). It reduces the amount of TRH secreted from the hypothalamus. TRH in turn stimulates the pituitary to produce thyroid stimulating hormone (TSH or Thyrotropin) and TSH stimulates the thyroid to produce the hormone T4.
T4 is an inactive metabolite that is reduced to T3 in peripheral tissues. T3, the active thyroid hormone is what you call a metabolic regulator. It is known to increase metabolism, thus burning more calories. Use of ephedrine, a stimulator of NE release has shown elevated levels of T3 for up to 12 weeks (8). This is most likely mediated via the reduction in NPY.
So in essence, all the adrenoreceptor exert some type of positive influence on fat loss. Only the BAR's however are uniquely pro-fat loss. The AAR's also exert negative effects. The reduction of these negative effects without compromising the positive effects will be an important part of fat loss supplementation.
Thyroid Hormone
Thyroid Hormone or T3, may also aid in fat loss, eventhough long term caloric restriction lowers T3 levels. So obviously, this is one target for supplementation that will prove a highly synergistic target.
The primary way in which T3 promotes fat loss, is by raising metabolism. That means using more calories to achieve the same. If you recall we discussed something similar in part 1 when we discussed BAT thermogenesis. Well, then it should be of no surprise that T3 predominantly works by increasing levels of uncoupling proteins (UCP's) that uncouple ATP synthesis from mitochondrial oxidation.
The first site of action is obviously UCP1, which is uniquely expressed in BAT and leads to an increase in thermogenesis. There are however two other UCP's, namely UCP2 and UCP3. UCP2 is most widely expressed, and UCP3 is only expressed in BAT and muscle tissue (10). The role of UCP3 is however not quite clear, as it plays no role in thermogenesis (9), bringing into question whether or not it is really an uncoupling protein.
Another method in which T3 increases fat loss is via its metabolite T2. T2 acts directly on the mitochondria to increase their productive ability and thus producing extra ATP, only to end up wasting it. An increase in ATPase activity ensues, thus breaking down the ATP again. A lot of it is wasted on something called 'substrate cycling' as well, which is the process of lipolysis, followed by lipogenesis.
Thus releasing and re-esterifying fatty acids. This creates a futile cycle that wastes ATP. This can be partially useful if the re-esterification can be inhibited. Most of the extra ATP however is wasted by increased heart rate. T2 increases the need for oxygen, so you take up more oxygen which leads to increased cardiovascular pressure to transport all that oxygen to where it is needed (11). BAT, coincidentally, is one of the largest consumers of extra oxygen (12).
T3 Molecule Closely related to the substrate cycling is the effect T3 has on BAR's. During long term stimulation by ligands, such as NE, phosphorylation and deactivation of the B1AR and B2AR can occur, leading to reduced lipolysis in WAT. T3 can increase the expression of BAR's (13) and increase your beta-adrenergic capacity.
Presumably this has something to do with substrate cycling, since increased BAR's stimulation would lead to more Adenylate Cyclase activity, which is also sort of an ATPase and can waste extra ATP. In this case much to our benefit, as we would be releasing fat.
It may also have to do with increasing thermogenic capacity, since T3 promotes the half-life time of the B3AR, which would further increase UCP1 expression and mitochondrial uncoupling (16)
Somewhat contradictory are T3's effects on insulin and insulin sensitivity. It seems to promote adipogenisis, not only via re-esterification of fatty acids, but also through increased sensitivity to insulin (14,15) . It is therefore wise to couple the manipulation of T3 to the manipulation of insulin sensitivity (cfr part 2).
However, hyperthyroid states generally lead to a reduction of insulin release (17), possibly due to increased apoptosis of the pancreatic beta-cells (18). This would make the effects on insulin roughly status quo I imagine.
T3 is also a Phosphodiesterase inhibitor (21). If you recall from article one, the A1AR stimulation lead to a Ca2+ dependent increase in PDE expression and PDE increased the breakdown of cAMP and the release of the inhibitory factor adenosine. Since T3 can reduce PDE somewhat, NE downregulates its own negative feedback by increasing T3 levels initially.
This same study showed that T3 can prevent Ca2+ dependent proteolysis and possibly spare muscle mass on a diet. This is however highly conflicting information, since T3 is only upregulated at the beginning of a diet, when you are less likely to lose muscle and of course the fact that T3 itself can exert a negative influence on muscle mass retention, since it initiates ubiquitin-proteasome related catabolism (13).
T3 also seems to increase Growth Hormone levels (22). But we will discuss the role of growth hormone in a future article
And then there is of course the question of whether or not T3 reduces A2AR (19,20) density. I'm inclined to believe it does, since increase in thyroid hormone usually leads to an increase in appetite, and it makes sense that T3 has several negative feedback channels, one of them possibly being an increase in NPY through negative regulation of A2AR. This would imply a dual action of T3, both negative and positive to fat loss.
The downside to T3 is that it severely stresses the heart, is catabolic to muscle, increases insulin sensitivity and appetite. These are some things that must be taken into account when trying to manipulate T3 levels in a diet. You want to prevent a drop in T3, but not necessarily increase T3 a whole lot.
Ok, we are nearing the end of our theoretical ordeal. Today we are going to be covering, briefly, Growth Hormone, appetite regulation, cortisol and TNF-alpha and how they may relate to our fat loss efforts.
This article is considerably easier than the previous three (part one, part two, part three), because it doesn't go quite as in-depth as the other three.
This wasn't necessary, since a cursory explanation of their actions is more than sufficient to understand their role in the physiology of fat loss. There are however, yet again, references to the previous installments, so those who have read up again have a plus, and will be able to follow better when we reach the less theoretical and more practical part of this series.
Growth Hormone
Growth Hormone is not exactly a major player in fat loss, but it can make a noticeable difference. In obesity we see that GH secretion is largely impaired, conditions where fat loss is enhanced, like starvation, usually display a significant increase in Growth Hormone. There is more than one reason to believe growth hormone may at the very least be a significant player in adipose tissue regulation, and vice versa, how adipose tissue is a regulator of Growth Hormone.
 The most obvious way in which growth hormone would aid in fat loss is of course its well-known effect on reducing insulin sensitivity. Growth Hormone has long been a popular product among pro's and amateurs alike, injecting it in the hopes of increasing IGF-1 levels and activity, and so increasing muscle mass. A very costly and largely ineffective waste of time, since the GH/IGF-1 axis is very tightly regulated.
The most obvious way in which growth hormone would aid in fat loss is of course its well-known effect on reducing insulin sensitivity. Growth Hormone has long been a popular product among pro's and amateurs alike, injecting it in the hopes of increasing IGF-1 levels and activity, and so increasing muscle mass. A very costly and largely ineffective waste of time, since the GH/IGF-1 axis is very tightly regulated.
But many have noticed the positive benefits of GH on body-fat reduction. Its mainly from this use however that we know GH increases insulin resistance. To such a degree that bodybuilders originally started supplementing with insulin to overcome this issue. The reduction in insulin sensitivity (cfr part 2) concurrently decreases adipogenic markers in fat cells, like PPARgamma and c/EBPalpha, creating a more suitable environment for fat loss.
But GH also acts directly on the fat cells to stimulate lipolysis. It does this both by inhibiting uptake of fat by fat cells, and increasing the release of fatty acids by those same cells. The primary regulator of uptake in the cells is Lipoprotein Lipase (LPL). LPL is secreted by the cell, removes fatty acids from tricglyceride portions and transports them inside the cell, where they are re-esterified.
GH has been shown to reduce LPL (1), possibly this is mediated by its effect on insulin sensitivity since insulin is primarily responsible for the translocation of LPL. The reduction may be greater in visceral fat (2), which may be the true benefit of Growth Hormone, since another lipolytic hormone, cortisol (discussed later) is extremely effective at reducing fat, but it increases visceral fat, something that could be inhibited by Growth Hormone manipulation.
Likewise it also stimulates the release of fatty acids from the adipocyte via methods discussed previously in articles 1 and 3 respectively. The first means by which Growth Hormone stimulates fatty acid release is by increasing the density of beta2 adrenoreceptors (B2AR) (for functioning of B2AR see part 1) much like thyroid hormone dose (cfr part 3) (3).
More B2AR means more effect of epinephrine and norepinephrine, stimulating amongst other things, lipolysis (release of fatty acids from white adipose tissue). A second manner in which Growth Hormone increases fat loss is also similar to Thyroid Hormone, namely by inhibiting Phosphodiesterase's (3).
As we saw in the functioning of the A1AR, norepinephrine also stimulates calcium signalling which results in enhanced PDE's. These increase the release of adenosine, and inhibitory factor for lipolysis.
An often confusing factor in using Growth Hormone for weight loss, is the fact that GH stimulates the release of IGF-1 under the right conditions. As the name suggests, Insulin-Like Growth Factor exerts insulin-like effects, increasing adipogenic markers (using the same pathways as insulin but a different receptor) and thereby promoting adipose tissue proliferation and differentiation.
GH itself has the opposite effect. Make no mistake, under caloric restriction IGF-1 is not increased. Its not exactly reduced either, but we do see distinct changes in regulators of IGF-1 activity, the IGF-binding proteins such as reductions in IGF-BP5 and increases in the negative regulator IGF-BP4. So the adipogenic effect of GH is pretty much completely reduced, allowing full expression of its lipolytic qualities.
GH manipulation makes a lot of sense during a diet as well. Increased serum fatty acids inhibit GH release, especially in the beginning of a diet that can be a problem. So increased GH release would be a definite addition. The effect on insulin resistance is also practical in the beginning, when calories are only reduced slightly.
Of course one can question the effect of whole-body insulin resistance in terms of muscle retention long term. In any case, practical application of GH therapy for obesity has demonstrated minor successes, so while it may not be the primary target for most of us, it is something worth considering.
Reduction Of Appetite
For a long time, the most important target for weight loss from a medical perspective, was a reduction in appetite. Makes sense, in animals weight was largely predicted by feeding behaviour. But then we seem to forget that animals don't eat refined foods like we do. Our foods are highly addictive and we don't just stop eating.
And a reduction in appetite does not correlate with a reduction in weight for most either. On top of that, cessation of treatment often resulted in a massive relapse. That's why weight loss from a research perspective, like we in the bodybuilding community do, focuses largely on promoting the actual release and burning of fatty acids, rather than addressing appetite.
 Appetite is important, but long term efficient weight loss needs to be paced. A severe decrease in appetite makes it hard to keep eating frequently, and if you don't eat frequently enough, your metabolism slows down and you hit a wall. Likewise too much appetite makes it hard to stay on a hypocaloric diet. So a reduction is highly beneficial, but not too much.
Appetite is important, but long term efficient weight loss needs to be paced. A severe decrease in appetite makes it hard to keep eating frequently, and if you don't eat frequently enough, your metabolism slows down and you hit a wall. Likewise too much appetite makes it hard to stay on a hypocaloric diet. So a reduction is highly beneficial, but not too much.
Most wintered dieters will also tell you that some sensation of hunger is necessary to keep yourself convinced you are still on the right course and to prevent any problems associated with dieting before they become too grave.
As a rule anything that aids in fat loss and helps a little along the way in terms of reducing appetite is a nice addition, but we rarely if ever employ products specifically designed for appetite reduction. That's definitely a positive thing, health wise.
Most beta-adrenergic drugs stimulate a reduction in the appetite, especially those that result in stimulation of the alpha receptors (5) (A1AR and A2AR) because these will lead to a decrease in orexigenic peptides, those peptides that usually make us hungry. The A1AR is obviously a more likely target, since it works somewhat pro-lipolytic at a local level as well, whereas the A2AR works anti-lipolytic at a local level.
Sufficient use of A1AR stimulators could allow you to more successfully employ A2AR blockers, although this may still result in a reduction of B3AR density (4) when used systemically. Drugs like ephedrine and methylphenidate are very potent reducers of appetite.
Cortisol
Cortisol is that one hormone we all love to hate, because its one of the most important inducers of proteolysis, resulting in muscle loss. Like most hormones we want some of it, but too much becomes detrimental. When we diet especially, cortisol levels tend to rise drastically. This is not entirely negative though, cortisol is probably the most powerful lipolytic drug there is.
 With a few downsides of course. Whereas it reduces fat in most areas, it increases visceral fat mass (beer belly like). This is an evolutionary safe-guard. Visceral fat can be easily mobilized, so relocating fat there may be to our advantage in surviving long spells of caloric restriction.
With a few downsides of course. Whereas it reduces fat in most areas, it increases visceral fat mass (beer belly like). This is an evolutionary safe-guard. Visceral fat can be easily mobilized, so relocating fat there may be to our advantage in surviving long spells of caloric restriction.
This is generally not a problem on a diet, visceral fat mass is reduced pretty much as fast as cortisol can relocate the fat there, especially as your diet progresses. The main negative is that if you fall off the wagon, and start binging, chances are you will gain fat in your gut first. The other major downside of course is the muscle loss.
Cortisol is the one hormone everyone would like to be able to control selectively. Turning it off in muscle and visceral fat, and turning it up in subcutaneous fat. This is why most steroid users will opt for an androgen with anti-cortisol properties to aid in retention of muscle mass.
Testosterone and trenbolone are the two most potent drugs in this regard, and they are highly synergistic in this regard as well. Testosterone blocks the cortisol receptor, whereas trenbolone may reduce receptor number and may reduce size of the adrenal gland long term.
Usually in a diet we try to make use of cortisol without letting it get out of hand.
TNFalpha
TNFalpha (Tumour necrosis factor alpha) is a cytokine that is not that different from cortisol in the way it acts. It also initiates catabolism in most tissues and has definitely been established as a factor that increases fat loss (6), but here too it's largely a choice between the positive and negative effects, because TNFalpha also reduces calorie expenditure.
It has been shown to reduced UCP1 (7) and UCP2 (8) activity, and with that, logically, a reduction in the amount of calories burned and in thermogenic activity. This may be, at least partially, due to its effects on catabolism in tissues rich in UCP1 (9) and UCP2.
PPARgamma negatively regulates TNF-alpha, so odds are when we interfere with PPARgamma we may be increasing TNF-alpha. Since PPARgamma increases the number of cells, and TNF-alpha reduces them, that makes perfect sense.
TNF-alpha closely relates to cortisol. For example it does make more cortisol available in cells. When glucocorticoids are excreted they are usually inactive. They need dehydrogenase enzymes to activate them. The 11-beta HSD enzyme family has been shown to modulate cortisone to create active cortisol. TNFalpha seems to increase intracellular activity of at least one of the 11-beta HSD enzymes (10). This is, in part, how it regulates its proteolytic and apoptotic effects.
Norepinephrine induced fat loss seems to protect against the apoptotic effects of TNF-alpha (11) however. TNFalpha represents a different means of non HSL-dependent fat loss, that results in permanent destruction of fat cells. This may be beneficial in extremely obese subjects, but the predominantly negative effects of TNF on muscle tissue make it a less likely candidate for dieting athletes.
TNF will be pretty much a non-factor when discussing dieting aids. It can be perceived as positive (increased fat loss) or negative (increased muscle loss) when a fat loss aid also increases TNF levels.
We have arrived at part 5, the last of the purely theoretical parts. After this we will be discussing more directly relevant topics such as actual ingredients and supplements that can aid fat loss. Of course a lot will depending on your understanding of these first 5 articles. This time around we are discussing the various sex hormones like DHEA, testosterone and estrogen.
Although not the main contributors to fat loss, they do play a crucial role in where fat is stored and how much fat is gained under anabolic conditions. Not to mention they play a pivotal role in the retention of muscle mass while dieting, which is still a major concern for most of us.
Apart from that we will also be discussing a little known factor, called adipsin, and leptin. The discussion of leptin is kept to a minimum however, since it will not be a major factor at all. It is only addressed here since it is often raised in discussion, and I do want to make this series a complete one.
Testosterone
Testosterone is a hormone with a dual effect. Partially lipolytic and partially adipogenic, or at least anti-lipolytic. This is clearly evident in the fact that it tends to decrease fat in older men, with low testosterone levels, but is largely seen as a steroid more likely to cause fat gain among athletes.
That testosterone is anti-lipolytic is somewhat logical. Androgens tend to increase IGF-1 release (2), which results in lower Growth Hormone and more activation of adipogenic markers. But it also exerts more direct effects. High levels of testosterone lead to higher levels of adrenoreceptors (3) in fat cells, and while testosterone does increase beta receptor density, it seems to have a predominantly pro-alpha anti-lipolytic effect (4).
On the other hand testosterone may exert a positive central effect, where it reduces appetite by lowering Neuropeptide Y, and increase Cocaine and Amphetamine related transcript (CART) (5) and has been shown to lower Lipoprotein Lipase (LPL) (6) which would in turn reduce the uptake of fatty acids into the fat cells.
Testosterone's main contribution with regards to body-fat is not so much its reduction, as it is the prevention of gaining additional fat mass in a pre-dominantly adipogenic (anabolic) environment. In the body we have pluripotent stem cells, that could develop into either pre-adipocytes or muscle satellite cells. Under the influence of testosterone, these stem cells are more likely to evolve into muscle satellite cells (23). This not only decreases the capacity to form fat cells in the body, it also increases our capacity for muscular repair, regeneration and hypertrophy.
Addition or manipulation of testosterone during a diet is usually considered with the retention of muscle mass in mind. Greater levels of protein synthesis, as well as a reduction in the activity of cortisol leads to more muscle being retained in a more catabolic condition, such as exists on a hypocaloric diet.
Knowing what to expect is therefore important, in that sense that testosterone may initially and dose-dependent, decrease fat tissue, but may end up slowing it down in the end. It may also be worth considering that testosterone seems to burn fat more efficiently in intramuscular and deep adipose stores, rather than subcutaneously, where the most visible fat pockets are located (7).
Estrogen
Estrogen is the second factor people often consider. Especially steroid and prohormone users are often plagues with a serious misunderstanding of estrogen and its effects on adiposity. Most are convinced that estrogen increases fat gain or retards fat loss, when in effect the opposite is true. Estrogens, and especially estradiol (E2), is probably a more effective fat loss aid than is testosterone. Although like testosterone, it may have certain anti-lipolytic effects by increasing a2 adrenoreceptors in specific female patterning (harder to lose fat in thighs and butt).
First of all, estradiol also reduces LPL (6), just like testosterone does, so uptake of fatty acids in adipocytes is reduced. Apart from that, its effects can be divided in three categories. Its effect on insulin-related events, its effects on Growth Hormone and its effects on reducing appetite.
Estradiol can cause a reduction in weight, with only a minimal effect in insulin itself (8), but that does not mean it does not alter the body's reaction to insulin. Estradiol lowers insulin receptor number (9), and in very high doses even actual insulin sensitivity (10). It does so in various ways, not in the least by reducing GLUT4 recruitment and translocation in adipocytes (11), which results in less glucose uptake in fat cells.
This will result in a negative energy balance and a greater activation of lipolysis, right where we want it, in the fat tissue. The effect of estradiol on insulin is quite acute, and clearly evident in the fact that short-term modulation drastically reduces glucose appearance (release) and disappearance (uptake) (12), suggesting a dysfunctional glucose transport system.
The second way in which estradiol may increase fat loss, is its effect on growth hormone (13,14). Unlike testosterone, which stimulates the GH/IGF-1 axis, the effect of estrogen may actually be in reducing systemic (liver-derived) IGF-1 (13), which lowers inhibition of Growth Hormone.
In doing so it obviously reduces the anabolic capacity of the body (which is why we don't use estrogen to build muscle) but increases the fat burning capacity since whole-body IGF-1 is reduced, leading to a reduction in adipogenic markers (since IGF-1 and insulin activate the same cascades) and a concurrent increase in Growth Hormone, leading to further decreases in LPL and upregulation of beta-adrenoreceptors (cfr. Part 4). Estradiol may even reduce IGF-1, while increasing IGFBP-3 (15).
This may in effect be the reason why estradiol does not promote growth, since unbound IGFBP-3, which is under normal circumstances the main carrier or IGF-1 in circulation, has been attributed charachteristics that inhibit growth (16). It acts as a pro-apoptotic agent to activate cysteine proteases, much in the same manner that cortisol or TNFalpha would (cfr. Part 4).
This implies that as long as we are seeing an increase in estradiol accompanied by an equal or larger increase in testosterone, we are reaping positive effects, on both fat loss and muscle retention since testerone increases IGF-1, while estradiol prolongs the half-life and effect of the hormone by increasing IGFBP-3 and IGF1-receptor density (20). Without the testosterone increase, it may however increase muscle loss (and potentially increase fat loss further by enhancing apoptosis of fat cells ?).
A third way in which estradiol helps as a fat loss agent is by reducing appetite. It reduces sensations of hunger via modulation of melanin-concentrating hormone. We have discussed the role of orexigenic (hunger inducing) peptides once or twice previously, specifically NPY.
Obviously NPY isn't the only peptide involved. For instance Agouti-related peptide is also involved, as is melanin-concentrating hormone (MCH). When energy intake is restricted, MCH levels sky-rocket, leading to an increased sense of hunger. Estradiol was able to completely abolish this increase in MCH (17), making it a very potent appetite suppressor during low-calorie diets.
Lastly, estradiol increases both the release of arachiconic acid (18) and the actions of cyclo-oxygenase (19) in certain cell types. This results in a quick and effective increase in several prostaglandins, including PGF2 and PGI2, that are related to lower body-fat levels. Because these effects can be highly varying in different cell types it should not automatically assumed that these events do occur, or that they necessarily contribute to fat loss however.
Estradiol can also prevent muscle loss, once again only in the presence of testosterone, by blocking the low affinity glucocorticoid receptors (22), protecting against the effects of cortisol. Testosterone, or another blocker of the high affinity receptors must be present however, otherwise the blocking of the low affinity receptor would not yield very good results.
On a closing note, as with testosterone, the effects of estradiol are not uniformly positive. It has been shown to enhance PPARgamma (20), so modulation of testosterone/estradiol levels should occur in the presence of a PPARgamma blocker for maximal effects on fat loss. And lastly, estrogen increasing products are often omitted during diets for the simple reason that estradiol increases aldosterone (21), a hormone that increases sodium retention, and as a result water retention.
Excess levels of estrogen often lead to water retention and a puffed up look. While this does not affect fat loss one iota, and can be addressed quickly, in only 1 or 2 days, it does make it difficult for the dieter to judge his progress accurately.
Adipsin
Once, when discussing the relevance of insulin resistance I commented that this would also reduce the esterification of fatty acids into fat cells. Someone then remarked to me that fatty acids stimulate their own esterification by increasing release of Acylating stimulating protein (ASP). But a lower insulin environment is conducive of more norepinephrine release and effect. When fat cells are stimulated by norepinephrine, they reduce their production of adipsin (1).
Adipsin (aka complement factor D) is a serine protease secreted by adipose cells (1). Adipsin can cleave complement protein C3 into C3a and C3b. C3a can be inactivated to C3adesArg, and C3adesArg is simply another name for ASP. So sympathetic activation of fat cells reduces their adipsin release, which reduces expression of ASP and consequently inhibits uptake of glucose and esterification of fatty acids.
This too makes perfect sense, when insulin is low, glucose uptake is low and that means a greater need for free fatty acids. It makes no sense to esterify them if they will be needed again shortly. Because they are not being esterified they can be transported out of the cell again and be burned.
DHEA
 Another hormone that should be well known to most is Dehydroepiandrosterone, or DHEA. It emerged as a supplement a while ago now, and was at that time hyped as another holy grail. It has since somewhat disappointed as a supplement, rarely being worth the money for most. Nonetheless new things are being uncovered about DHEA on an almost daily basis that do really prove it plays a key role in many bodily functions.
Another hormone that should be well known to most is Dehydroepiandrosterone, or DHEA. It emerged as a supplement a while ago now, and was at that time hyped as another holy grail. It has since somewhat disappointed as a supplement, rarely being worth the money for most. Nonetheless new things are being uncovered about DHEA on an almost daily basis that do really prove it plays a key role in many bodily functions.
As an intermediate step to the production of testosterone and estrogen, as a neuroactive steroid and behavioural regulator and … as a lipolytic substance. Surprisingly DHEA was never marketed as a fat loss drug. In contrast, an analog called 7-oxo-DHEA WAS marketed as a fat loss drug.
In direct comparison however, the use of DHEA resulted in a much lower fatty acid content in fat cells, while 7-oxo-DHEA actually INCREASED fatty acid content in the fat cells (24). The difference correlated highly with the difference in stearoyl-CoA desaturase (SCD1).
Most likely part of the difference is mediated by DHEA's effect on SCD1. Another possible contributor is the fact that 7-Oxo-DHEA reduces the formation of active cortisol. While this may be somewhat effective in regards to minimizing loss of muscle, it is a huge negative with regards to fat loss, where cortisol is highly lipolytic in all tissues safe for visceral fat. Athletes rarely if ever have a high visceral fat depot, and if so, with training it is the first fat depot likely to get reduced since it is in a position to be readily used by the liver. Making visceral fat the least of the concerns of the exercising individual.
Furthermore it was concluded that DHEA, but not 7-oxo-DHEA, reduced the differentiation of pre-adipocytes to full adipocytes. Preventing actual fat gain, under the influence of hormones that increase differentiation, such as leptin, IGF-I and insulin.
Most likely this occurs through the reduction of expression of PPARgamma (25). And on a final note, that same study also proved that in differentiating pre-adipocytes, DHEA promoted thermogenisis (formation of brown fat cells) while 7-oxo-DHEA had no effect on thermogenisis at all. This is corroborated by a study (26) showing that DHEA increases expression of UCP1 and UCP3 in brown fat cells. Since the expression of UCP1 technically makes a fat cell a brown fat cell, this means DHEA promotes the formation of BAT over WAT.
As if this were not enough, DHEA also increase beta-adrenergic fat loss (27) through a mechanism that does not involve the reduction of adenosine activity. That means it will again support any and all drugs that use the c-AMP dependent cascade to releasing fatty acids.
Meaning that DHEA increases fat burning, decreases fat storage, fat cell formation and of the fat cells that do get formed, it promotes the formation of those that can contribute to fat burning. That makes DHEA probably one of the most versatile hormones in the body with regards to fat loss. Products that affect DHEA positively, would also affect fat loss positively therefore.
Leptin
Leptin is a hormone that is primarily secreted from the fat cells themselves, in response to the amount of triglycerides they stock. On a paracrine level they serve to signal neighbouring pre-adipocytes that the existing fat cells are full and that the pre-adipocytes need to proliferate and differentiate.
It does so via a specific leptin receptor that uses the same downstream signalling cascades as insulin, and results mostly in similar effects, namely the activation of cEBP alpha and PPARgamma, factors of proliferation and differentiation, causing an increase in the amount of fat cells.
Because leptin serves as a signal of excess, it fulfils many functions on an endocrine level that may be perceived as positive to fat loss, such as a reduction in appetite, diminished response to appetite inducing signals, and so forth. Mainly because of its many roles in this regard, it has been hyped as the next big factor in weight loss. Of course, anything is a big factor if you represent it on a very simplistic level.
When leptin becomes the dominant signal, it pretty much regulates everything. It lowers LH, it lowers TSH and it increases CRH. Normally this would lower testosterone and T3, and increase cortisol, resulting in more muscle loss and a slower metabolism, if it wasn't that leptin itself maintains these functions in the stead of the normal regulatory mechanisms. So far so good. Of course, unless you plan to inject leptin, your leptin levels will eventually drop.
As triglyceride count drops, so does the signalling thereof. Even with supplements proven to increase leptin, increases occur percentagewise, meaning they are much lower in individuals with lower tri-glyceride stores. The leaner you get, even with supplementation, the lower your leptin levels will get.
At a certain point leptin gets too low to exert much of an effect. But since all the time you spent trying to keep leptin elevated lowered your TSH and LH, and increased CRH, you are now left with nothing to regulate testosterone and thyroid hormones, resulting in a lower metabolism more prone to store fat, and at the same time increasing cortisol levels, which results in greater muscle loss.
One study (28) showed that people who experienced a faster and greater DROP in leptin levels, early in the diet, lost weight faster and kept it off longer. This caused me to postulate that lowering leptin faster might be a wiser course of action. I attempted and tested this by lowering all factors that could increase leptin (mainly vitamin E and zinc) without making any other changes. Indeed, progress occurred more rapidly than normal.
However I might have been equally erroneous in my attempt, since I learned that zinc may be adipogenic through its manipulation of IGF's, and that the increase in weight loss may therefore have been due to less IGF-I signalling, rather than a drop in leptin. I was equally foolish to believe that a manipulation in the other direction would pay off. Leptin, as it turns out, is a non-factor in the diet. This is of course my opinion, and many people are less than happy with this opinion. So make up your own mind.
What I can tell you is that leptin uses the same downstream signalling cascades as insulin, and has the same negative effects as insulin as a result, at least in adipose tissue. Since it is released there, it is also safe to assume its autocrine/paracrine effects are greater than its endocrine effects.
Moreover, even if it differentially manipulated these cascades, resulting in a positive effect, it would still be less of an advantage than simply reducing insulin resistance. Since this is a valid way to increase lipolysis, any and all attempts at manipulating leptin would be utterly futile anyway, since the body would lose its sensitivity to leptin. Most likely this has a lot to do with the fact why manipulating leptin negatively was just as nonsensical as manipulating it positively.
In people with Type II diabetes leptin is increased phenomenally, as is insulin. These people keep increasing their bodyweight, despite the homeostatic role of leptin. Again, this is as a result of the cross-signalling between leptin and insulin. An insulin insensitivity will automatically decrease leptin sensitivity, so the lack of effect here is as a result of insensitivity to leptin.
Such people may highly benefit from an increase in leptin sensitivity, as well as insulin sensitivity. Of course, since this targets the same cascades, treating one largely treats the other. If you suffer this type of insensitivity, or fear you may suffer it, it is in your own best interested to get this tested and to receive proper treatment for your condintion by a licensed medical professional, rather than to put your hopes in unregulated supplementation.
For non-obese individuals however, leptin remains largely, a non-factor due to the better results with insulin desensitizing drugs (rendering leptin inert), the difficulty in manipulating leptin naturally and the lower tri-glyceride stores.

 1. Canned Tomatoes
1. Canned Tomatoes

{kind=link}